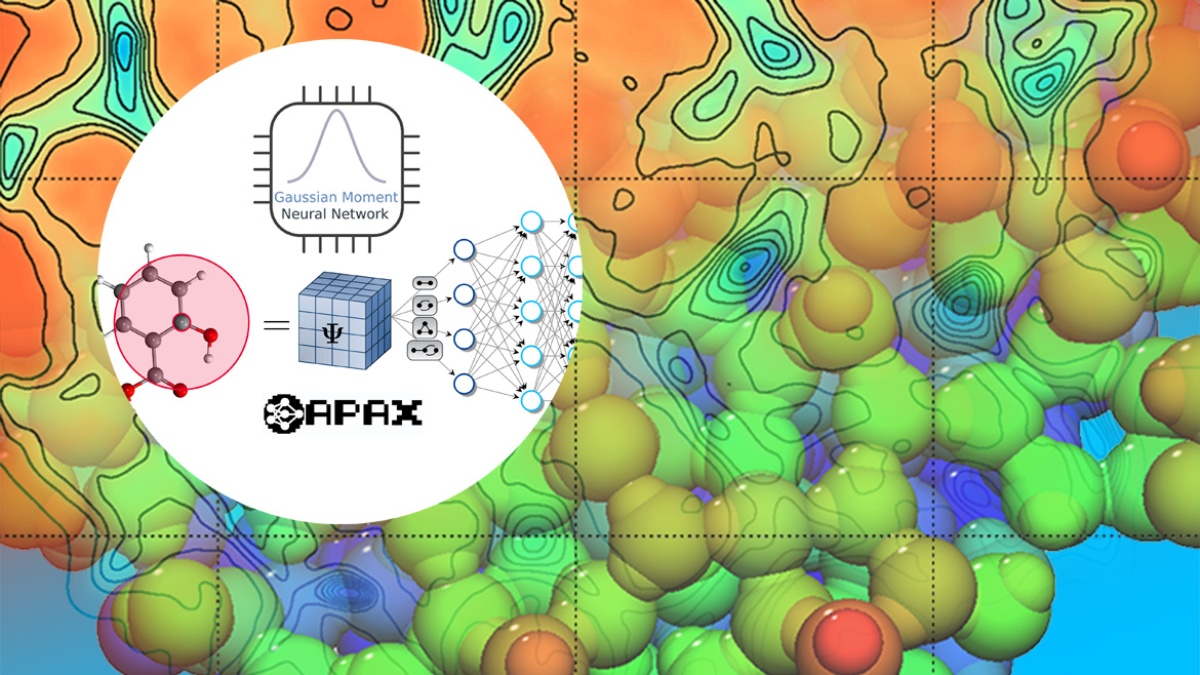
Neural networks and Gaussian process regression provide surrogate models for the potential energy surface. They can be evaluated much faster. It limits the required energy evaluations for tasks like rate constant calculations and geometry optimization.
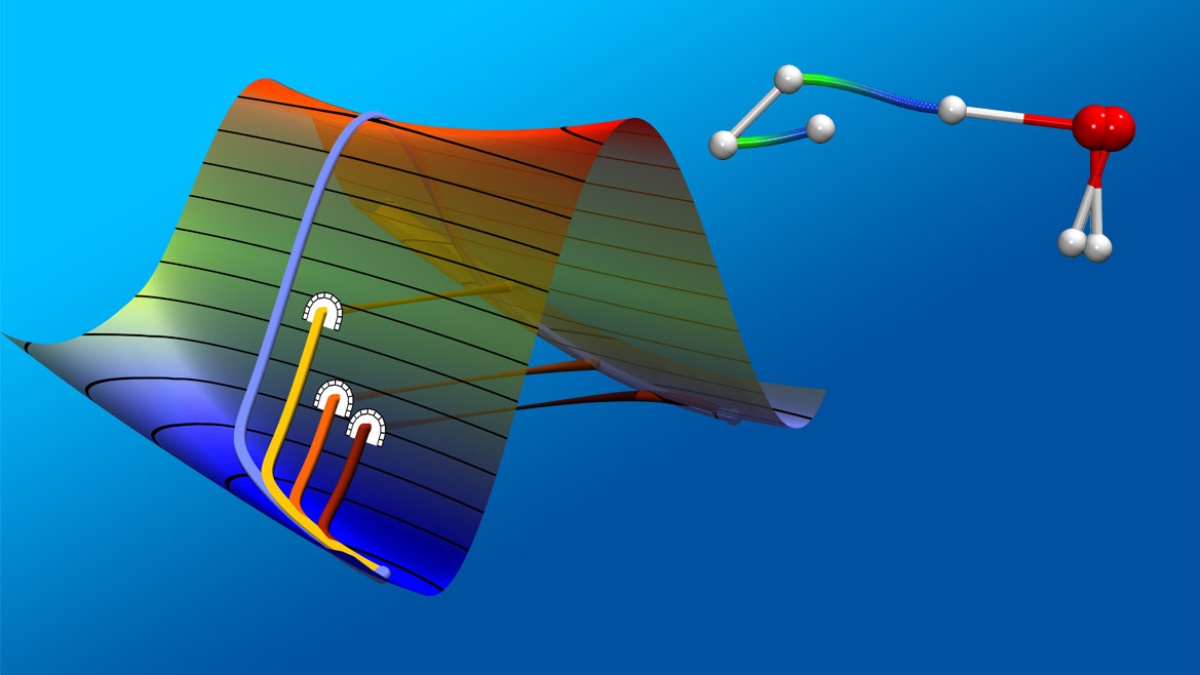
We develop methods around instanton theory to calculate tunneling rate constants in chemical reactions and implement them in our open-source code DL-FIND. This theory provides high accuracy at reasonable computational costs, even for systems as large as enzymes. Our work on tunneling was funded by the ERC.

The darkness readily observed between the stars on a clear night sky is far from empty. A large variety of molecules has been detected during the past century. We contribute with our rate constant calculations of atom tunneling.
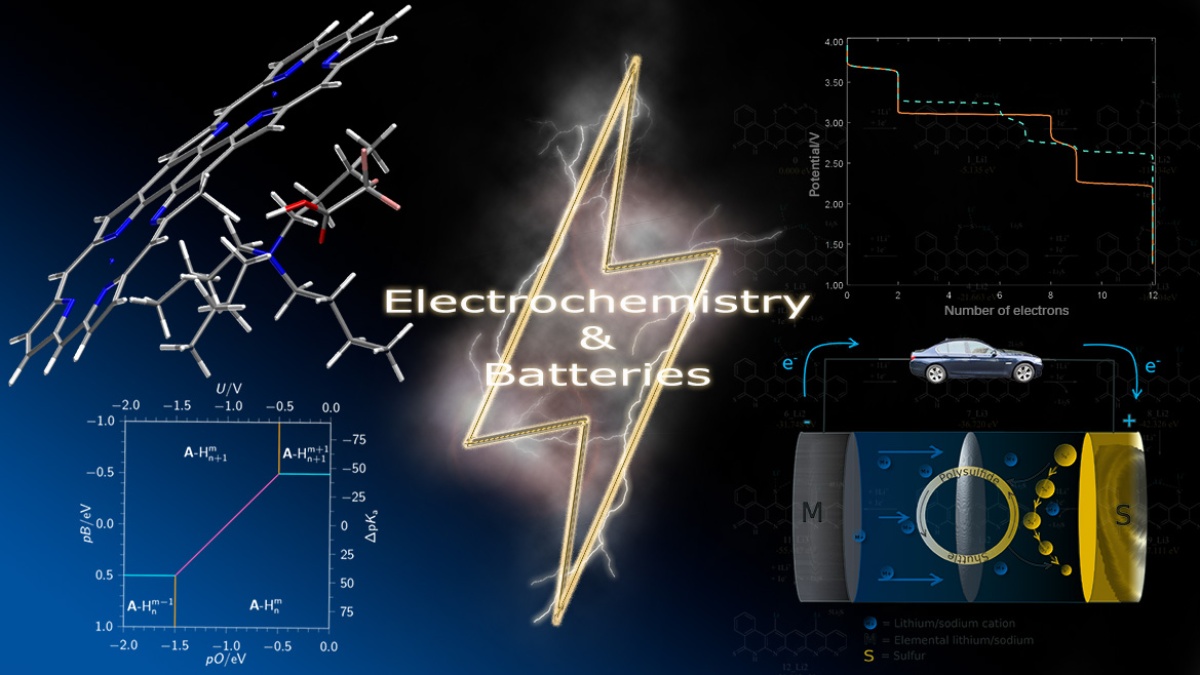
Electricity drives chemical reactions, and chemistry stores electricity in batteries. We aim to understand electrochemical processes occurring in electrolyte solutions using quantum chemistry as a molecular microscope.
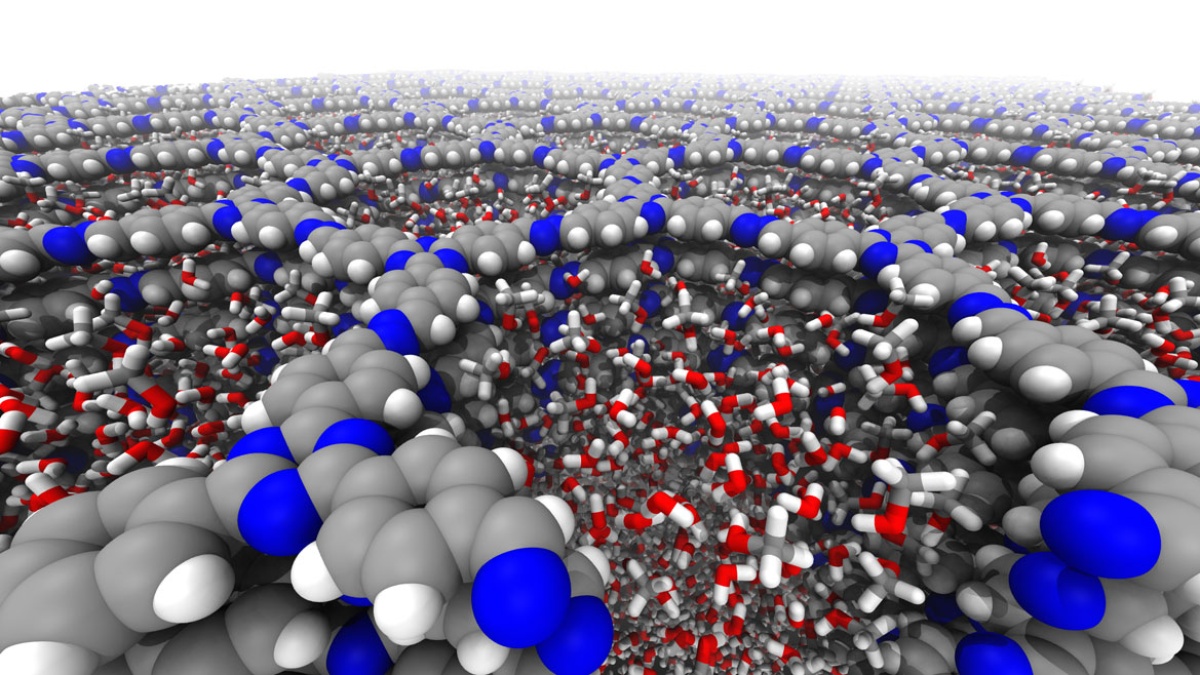
We investigate molecular heterogeneous catalysis in pores with quantum chemical and machine-learning techniques in cooperation with experts from organic and inorganic chemistry, analytical chemistry, and physics. The project is funded by the CRC 1333.
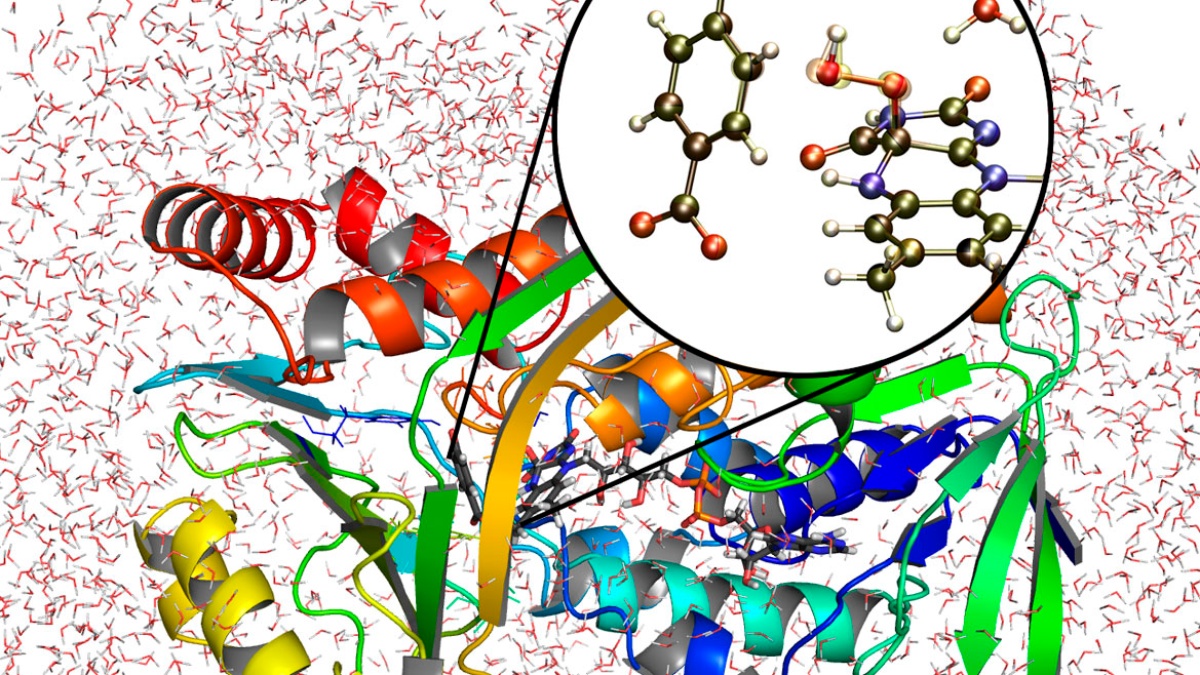
Transition-state search, rate calculations, and machine-learning-based optimization: The PI developed the modular geometry optimization library DL-FIND. It can be interfaced with atomistic simulation codes. We use it in conjunction with ChemShell. DL-FIND development is a collaboration with Daresbury Laboratory, UK.
The Computational Chemistry Group simulates chemical and biochemical reactions under consideration of environmental effects. We complement experiments and provide answers where experiments alone cannot do so. We study catalysis, surface processes, astrochemistry, enzymes, and materials.
Application areas:
- Catalysis
- Astrochemistry
- Surface science (CRC ATLAS)
- Electrochemistry, batteries
Method development:
We develop methods that allow us to investigate these kinds of problems in innovative ways. We use machine learning techniques to investigate chemical reactions and to optimize geometries. We co-author the program package ChemShell, and we lead the development of the optimization library DL-FIND. Our group extends the capabilities of geometry optimization in systems with many thousand degrees of freedom and developed methods to calculate tunneling processes in large systems as well as the free-energy sampling technique umbrella integration.

Main Funding Sources
- ERC Consolidator Grant TUNNELCHEM
Completed grant by the European Research Council - SimTech Cluster of Excellence
A project that brought the PI to Stuttgart and that keeps us inspired! - SFB 1333: Molecular Heterogeneous Catalysis in Confined Geometries
Opening new frontiers in catalysis - LILAC consortium in AstroChemistry
Astrochemistry requires close interaction with astronomical observation, lab astrophysics, and modeling - Artificial Intelligence Software Academy
Cutting-edge research and training in AI - SPP 2363 on Utilization and Development of Machine Learning for Molecular Applications – Molecular Machine Learning
AI and ML for molecular applications - SFB 1667: Advancing Technologies of Very Low Altitude Satellites
We find out what the remaining atmosphere in 200 km altitude does to satellites.